有时候构建的蛋白不起作用, 想要预测一下蛋白的结构进行检查. 可以使用AlphaFold(的变种)进行蛋白质结构预测, 并使用Pymol对蛋白质结构进行比较和可视化.
AlphaFold使用
AlphaFold已经开源了, 但是没有合适显卡的电脑运行起来会特别慢, 故这里首先想办法白嫖别人的服务器来跑蛋白质结构预测.我目前发现的能白嫖的服务器只有Google的Colab和Phenix项目的服务器, 故要先自行解决科学上网的问题, 如果要用Colab的话还需要注册一个谷歌账号. (有段时间北鲲云注册新用户时候会送代金券, 可以直接用于跑AlphaFold, 但是现在不氪金好像用不了了.)
Colab中AlphaFold的使用
Colab上常用的AlphaFold(衍生品)主要有:
- AlphaFold: https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb
- ColabFold: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb
二者预测效果相差不大, 但是ColabFold会快不少, 一般比较推荐选择ColabFold.
打开上述ColabFold页面, 登录Google账号, 随后输入待预测蛋白质氨基酸序列(query_sequence)和名称(jobname), 一般余下参数采用默认值即可运行(Runtime > Run all或者代码执行程序 > 全部运行), 运行完毕后找到 Package and download results部分下载模拟结果.
如果模拟结果中的蛋白质与数据库中的结构相差巨大, 可以考虑加入模板对预测过程进行指导. 将template_mode项置为custom, 随后运行整个程序, 在template_mode项后面可以找到文件上传入口, 上传模板文件后程序才会向后执行预测.
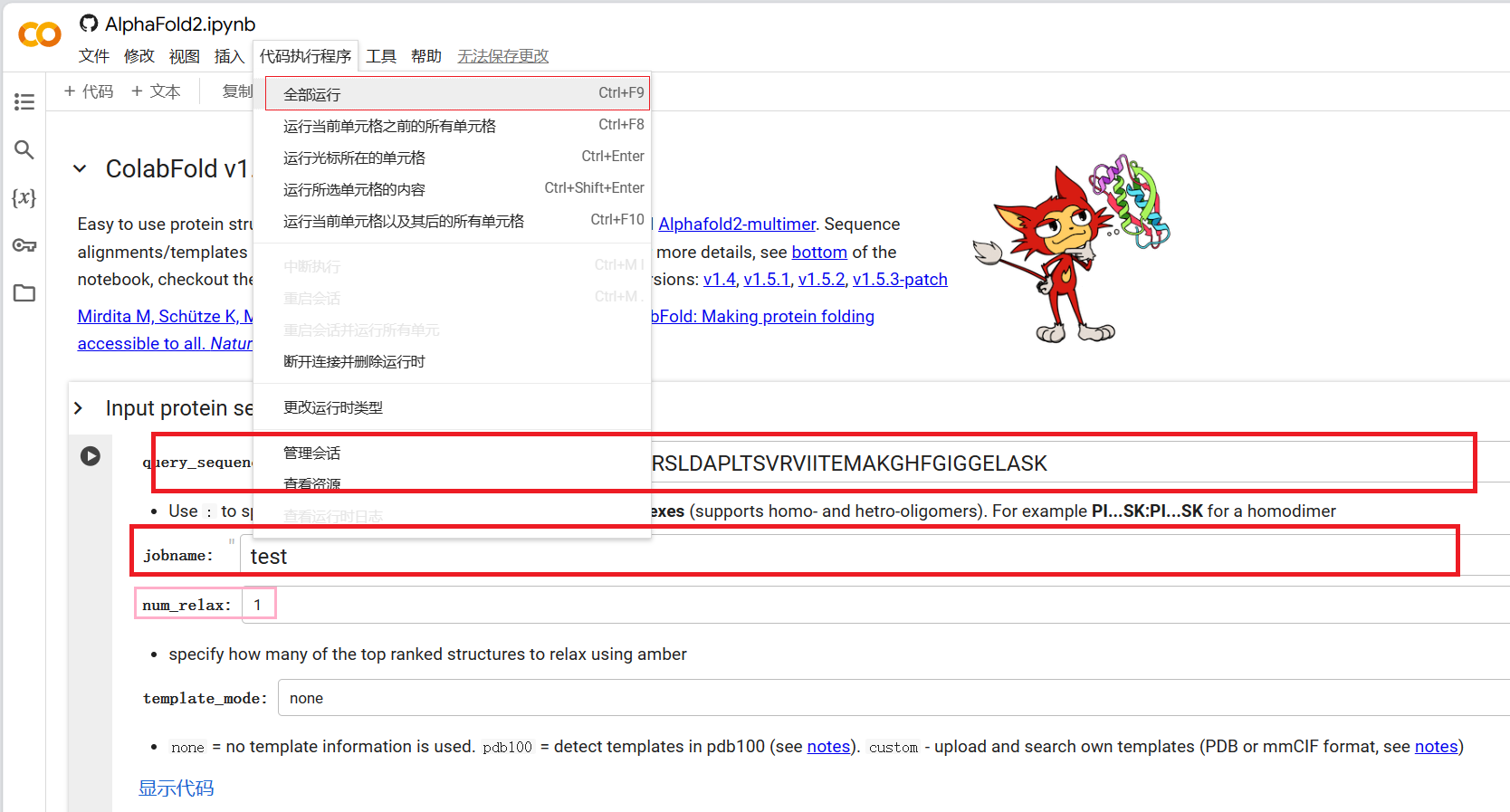
对于蛋白质复合体, 使用:隔开各亚基的氨基酸序列并全部填入query_sequence中即可. 有些人使用一段足够长的柔性linker连接复合体各个片段, 再输入ColabFold中进行预测, 我个人认为AlphaFold无规则卷曲的预测能力较弱, 在没有相似复合体模板指导的情况下这样得到的预测结果不一定可靠.
注意免费版Colab服务器在浏览器页面关闭或者连接断开后就会自己关掉, 就得重新去跑结构预测程序了. 同时免费版Colab会限制用户算力的使用, 刚跑过结构预测任务后数小时内内有概率没办法运行任何结构预测任务, 这时候要么付钱要么等一等再预测下一个蛋白的结构.
Phenix的安装与其AlphaFold模块的使用
免费版的ColabFold使用中有诸多限制, 故我将目光投向了更方便白嫖的Phenix, 这是一个处理大分子结构数据的软件集合.
Phenix的安装
Phenix需要在本地安装一个基于Python的客户端, 并使用客户端访问其服务. 安装流程见下:
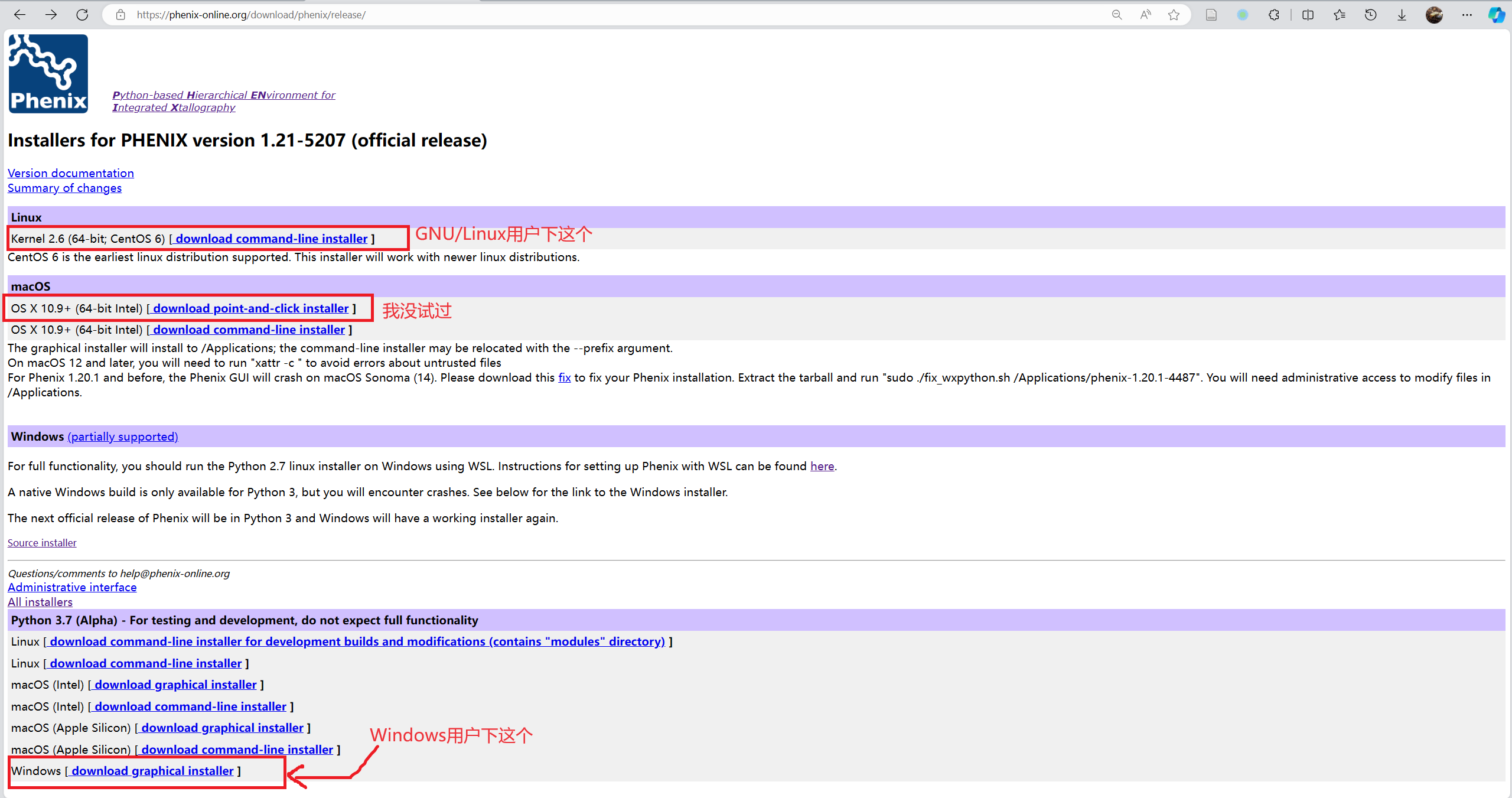
- 在https://phenix-online.org/download, 点
Request password for academic users, 使用学校邮箱申请下载密码. - 审核大概需要半个到一个工作日的时间, 会返回一封邮件说明用户名和密码.
- 回到https://phenix-online.org/download, 点击
Download official release, 输入用户名(download)和密码登录. - 根据自己的系统选择安装包下载, 安装包较大, 下载要是失败了就多试几次.
安装过程
Windows系统:
图形化界面的安装包, 一直点下一步就好.
安装完毕后找到Phenix程序目录(默认在
C:\Users\用户名\phenix-x.xx-xxxx), 在该目录下找到Library\bin\phenix.bat, 双击这个bat文件运行Phenix.通常情况下需要翻墙才能连接上Phenix的服务器, 为此新建文件
Library\bin\phenix-proxy.bat, 右键使用记事本编辑, 写入下述代码并保存.set http_proxy=http://127.0.0.1:7890 # 需要替换为自己梯子软件中的的参数, 此处为clash默认值 set https_proxy=http://127.0.0.1:7890 # 需要替换为自己梯子软件中的的参数, 此处为clash默认值 phenix.bat双击运行
phenix-proxy.bat会打开一个命令行窗口并运行Phenix, 如果希望隐藏窗口, 可以新建Library\bin\phenix.vbs, 右键使用记事本编辑, 写入下述代码并保存.set ws=WScript.CreateObject("WScript.Shell") ws.Run "phenix-proxy.bat",0双击
phenix.vbs即可运行Phenix, 如果希望在开始菜单中找到它, 可以将它复制并在C:\Users\用户名\AppData\Roaming\Microsoft\Windows\Start Menu\Programs下建立一个快捷方式, 重启后就能在开始菜单中找到.
GNU/Linux系统:
解压下载到的安装包, 并打开终端,
cd入解压目录,ls后应该能看到README文件和install文件等给
install文件运行权限:chmod +x ./install运行
install安装脚本:sudo ./install, 软件将会被安装在/usr/local下, 如果要自定义安装目录, 在前述语句后面追加参数成sudo ./install --prefix=/目标安装位置/的/绝对路径.打开终端,
cd入Phenix安装目录ls后应该能看到phenix_env.sh等文件, 运行source ./phenix_env.sh && phenix来打开Phenix. 如果遇到报错, 需要考虑系统是否安装完整英文的locale, Debian系的系统可以用sudo apt install language-pack-en\*来安装.通常情况下需要翻墙才能连接上Phenix的服务器. 运行下述命令设置代理并打开Phenix.
export http_proxy=http://127.0.0.1:7890 # 需要替换为自己梯子软件中的的参数, 此处为clash默认值 export http_proxy=http://127.0.0.1:7890 # 需要替换为自己梯子软件中的的参数, 此处为clash默认值 source /Phenix/的/安装目录/phenix_env.sh phenix
Phenix下AlphaFold的使用
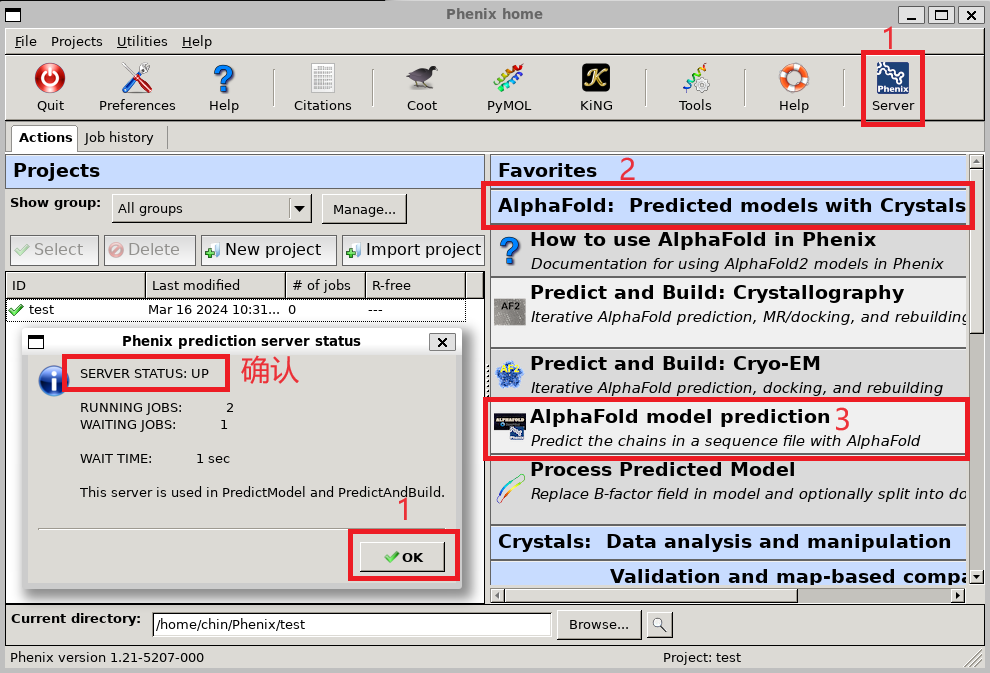
第一次打开Phenix需要新建一个Project, 随后进入Phenix主界面. 首先确认Phenix Server能够连接上, 然后打开 AlphaFold: Predicted models with Crystals or Cryo-EM > AlphaFold model prediction.
进入AlphaFold model prediction的界面后, 随便写一个Job title, 随后在Input files区域添加fasta格式的氨基酸序列文件, 软件会自动将序列提取后显示在下方Composition区域. 也可以直接在Composition区域输入或者修改氨基酸序列. 随后点击上方的Run即可运行预测.
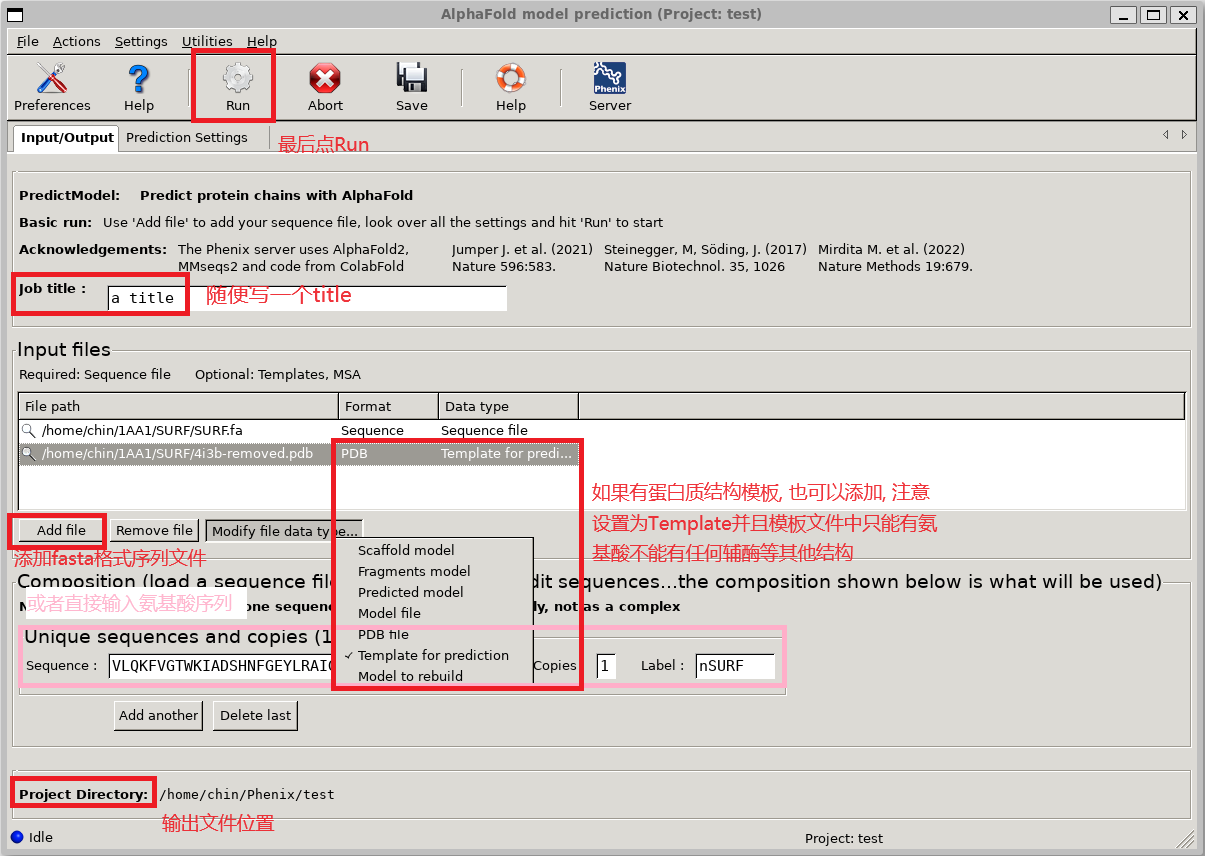
有时候对已知结构蛋白进行了突变, 想要预测突变后的蛋白结构, 可以在Input files区域添加原始蛋白的结构文件, 将其标记为Template for prediction后再进行预测, 通常能够得到更准确的预测结果. 预测融合蛋白结构时, 也可以通过提供各部分的已知结构作为模板, 来避免预测结果和已知结构偏差过大的情况. 注意: 直接从PDB下载的结构文件中可能包含有水/配体分子等非氨基酸基团信息, 这样的文件直接用作模板会导致phenix报错, 要先使用Pymol之类的软件删除这些分子的信息.
Phenix没有处理多聚体结构预测的能力.
LocalColabFold的安装与使用
直接使用AlphaFold的话需要下载大量的数据库, 下载缓慢并且占用大量硬盘空间, 故此处只尝试ColabFold的本地安装与使用. ColabFold将序列比对的任务交给专门的MSA服务器执行, 本地只需要有预测程序和模型文件即可. 已经有人将ColabFold的安装写成自动化脚本了, 几句命令就能完成, 详见https://github.com/YoshitakaMo/localcolabfold.
ColabFold在Unix like系统上都能运行, 包括各类GNU/Linux、Mac OS, 如果Windows系统要用的话得开启WSL. 此处以Ubuntu 20.04为例.
LocalColabFold的安装
Nvidia CUDA环境安装. 不安装CUDA也能用CPU死跑, 就是速度慢一个数量级. 不过实测即使安装了CUDA也有大概率软件识别不出来 在https://developer.nvidia.com/cuda-downloads/, 根据自己的系统查找合适的runfile后网页会给出现在安装指示, 照做就好, 例如:
wget https://developer.download.nvidia.com/compute/cuda/12.4.1/local_installers/cuda_12.4.1_550.54.15_linux.run sudo sh cuda_12.4.1_550.54.15_linux.run注意CUDA安装包较大, 如果下载失败(wget提示连接断开), 就删掉重新下载run文件. 安装时候如果报错, 可能是系统旧驱动没有卸载或者图形化界面运行阻止了显卡驱动的安装, 运行:
sudo apt purge \*nvidia\* -y和sudo systemctl stop gdm. 注意运行这些命令后系统的图形化界面将被关闭, 只能在原始的命令行界面中输命令继续进行安装操作.
有自动化安装脚本, 注意不要连接梯子来安装, 否则安装失败还没有报错, 但是后期程序运行不起来. 运行下述命令来安装.
sudo apt update
sudo apt -y install curl git wget build-essential
wget https://raw.githubusercontent.com/YoshitakaMo/localcolabfold/main/install_colabbatch_linux.sh # 这个脚本如果下不下来的话可以使用梯子下载或者把地址输入浏览器打开再另存为本地文件
bash install_colabbatch_linux.sh # 这个命令不能挂梯子运行
echo "export PATH=$PWD/localcolabfold/colabfold-conda/bin:\$PATH" >> ~/.*shrc
LocalColabFold的使用
首先要准备好待预测蛋白的氨基酸序列, 以fasta的格式写入一个文本文件. 如果是复合体, 使用:隔开各个亚基的序列. 如果要提供蛋白质结构模板, 将所有模板结构pdb文件放在一个文件夹里. 随后运行:
colabfold_batch --templates \
--custom-template-path /模板/结构/pdb文件/所在的/文件夹/路径 # 如果不打算使用自己的蛋白质结构模板, 就删掉这行
--amber --num-relax 1 \ # 如果只看得分最高的预测模型, 则没必要增大这个数字
--use-gpu-relax \ # 如果电脑没有独显则可删掉这句
--num-recycle 3 \ # 增大这个参数会另预测结果更精准但是延长预测耗时
--num-models 3 \ # 取值在1到5之间, 控制输出的模型数量
/待预测/蛋白/序列文件/路径 /输出/模型/存放/文件夹/路径
第一次运行时候需要下载AlphaFold模型参数, 耗时大约1 h, 如果下载失败就多试几次或者挂个梯子. 运行结束后在/输出/模型/存放/文件夹/路径查找预测的pdb文件.
Fasta格式举例:
>1BJP_homodimer PIAQIHILEGRSDEQKETLIREVSEAISRSLDAPLTSVRVIITEMAKGHFGIGGELASKVRR: PIAQIHILEGRSDEQKETLIREVSEAISRSLDAPLTSVRVIITEMAKGHFGIGGELASKVRR
Pymol的使用
其实ChimeraX也能完成Pymol完成的任务, 功能更加强大, 但是界面相对复杂, Pymol界面相对简单一些.
Pymol官方的版本是要收费的, 但是其源代码是开放的, 因此可以安装第三方编译的版本.
Pymol的安装
对于GNU/Linux系统, 软件仓库中大多已经有编译好的Pymol, 直接安装即可, 比如Debian系系统使用命令sudo apt update && sudo apt install pymol即可安装Pymol.
对于Windows系统, 操作略微复杂:
- 安装Python: 在Python官网下载安装包斌安装, 注意最后最好勾选类似”Add python.exe to PATH”的选项, 如果没有, 则需要手动添加(开始菜单搜索”环境变量”, 打开环境变量后找到”Path”, 在其中加入两条记录:
Python\安装\路径, 和Python\安装\路径\Scripts即可). 安装后最好重启电脑. - 安装Pymol依赖的Python程序包: 打开命令提示符, 运行
pip install numpy setuptools pmw pyqt5 - 下载Pymol程序包: 去Github搜索
Pymol Windows, 根据CPU架构和Python版本下载对应的pymol***.whl和pymol_launcher***.whl(可选). - 安装下载的whl包: 进入whl包所在的文件夹, 在地址栏输入
CMD回车即可进入命令提示符, 输入命令pip install .\pymol***.whl来安装相应的包, 同理安装pymol_launcher***.whl(可选) Python\安装\路径\Scripts下能够查找到pymol.exe, 双击即可运行Pymol. 如果前述操作中安装了pymol_launcher***.whl, 则会在Python\安装\路径下查找到PyMOL.exe, 同样可以用来打开Pymol.- 可以给上述的
pymol.exe或PyMOL.exe创建快捷方式并放到C:\Users\Chin\AppData\Roaming\Microsoft\Windows\Start Menu\Programs中, 这样重启电脑后就可以在开始菜单中打开Pymol.
Pymol的使用
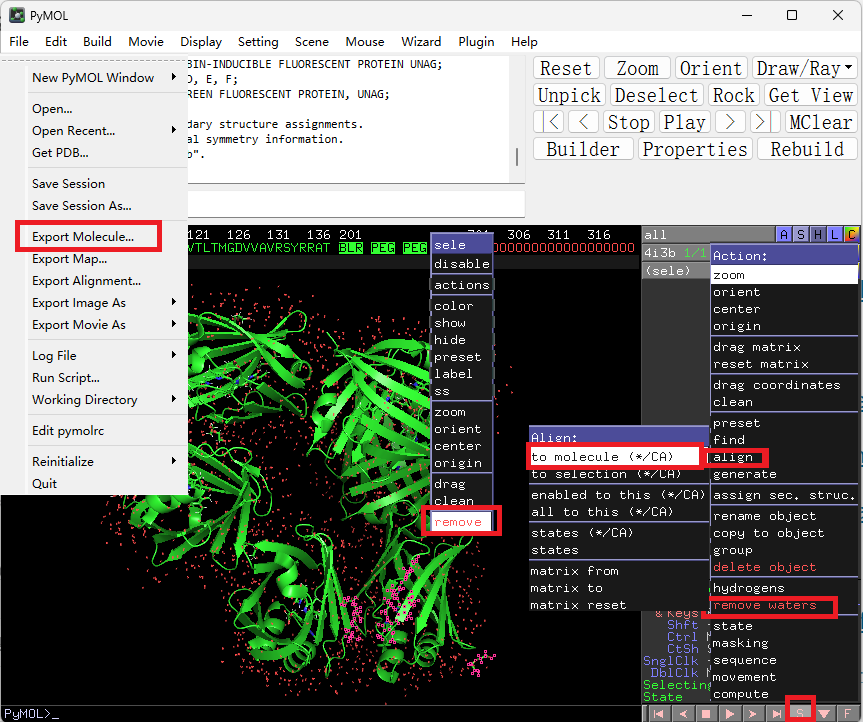
- 编辑PDB文件
PDB文件中若是有除了氨基酸残基外其他分子的信息, 输入某些AlphaFold变种作为模板时可能会报错, 此时需要将多余分子的信息删除. 将PDB文件拖入Pymol内, 点击右下角的S打开序列, 选中非氨基酸的部分右键再点击remove即可将相应内容删除. 如果要去除水分子的数据, 可以按下右上角的A, 在其中找到对应的功能remove waters. 注意不要直接保存, 那样会保存成Pymol专用的PSE文件, 需要使用File > Export Molecule...来导出为常见的PDB文件或者CIF文件.
- 对其结构相近的蛋白
预测好的蛋白质结构并不会与模板在同一个位置, 比较二者异同时候需要让二者尽可能重叠, 以凸显差异之处. 将多个PDB文件拖入Pymol中, 软件右侧会显示出多个文件名, 选择预测结构后面的A, 点击出现的Action菜单中的align > to molecule (*/CA) > 模板文件名, 即可将预测到的结构对其到模板结构上去, 同时会用细线标识出二者对应氨基酸之间的距离.
本博客所有文章除特别声明外,均采用 CC BY-SA 3.0协议 。转载请注明出处!